数据处理及画图
这是我们最重要、最耗时的步骤。下面的内容将以对WRKY和MADS基因家族的分析为例。
通过对他人文章的阅读,我们了解到这项工作一般需要完成以下的内容:
我们也可以做
识别并找出基因家族的基因
现在我们有完整的基因序列和蛋白质序列文件,如何从这些序列中找出所有属于某个基因家族的序列?
这一部分会啰嗦一些,会涉及软件安装的每一步,后面的部分不会这么详细。
目标
从基因组文件中找到并提取出我们需要的序列
材料与软件
- HMMER
- 编码蛋白质的fasta文件
主要步骤
- 明确要找的基因家族
- 下载HMMER软件
- 找文献,找hmm profile文件
- 从interPro网站上下载该基因家族对应的文件
- 使用HMMER搜索序列
- 从fasta文件中提取对应的序列
原理
HMMER利用隐马尔科夫模型来识别结构域,从而找出基因家族序列。
隐马尔科夫模型这是一个统计学上的概念,具体我也很难解释。这里试着体会一下
马尔科夫链是一个描述状态序列的数学模型。它的核心思想是 “无记忆性”,即系统在某个时间点的状态,只取决于它在前一个时间点的状态,而与更早之前的状态无关。假设我们用一个简单的马尔可夫链来模拟天气。我们设定天气只有三种状态:晴天、多云、雨天。
- 如果今天下雨,明天是晴天的概率可能很低,而明天继续下雨的概率会比较高。
- 如果今天晴天,明天多云的概率可能比较高。
这个状态之间的转换概率,我们可以用一个“状态转移矩阵”来表示。比如:
| (今天) \ (明天) | 晴天 | 多云 | 雨天 |
|---|---|---|---|
| 晴天 | 0.7 | 0.2 | 0.1 |
| 多云 | 0.3 | 0.4 | 0.3 |
| 雨天 | 0.1 | 0.4 | 0.5 |
这个模型很简单,因为状态(天气)是可以直接观察到的。
这时要开始“隐”了,例如我们看不到天气,但我们能知道某人是否去晨跑了。他去不去晨跑和天气有如下关系:
| 隐藏状态 (天气) | 跑 | 没跑 |
|---|---|---|
| 晴天 | 0.8 | 0.2 |
| 多云 | 0.7 | 0.3 |
| 雨天 | 0.1 | 0.9 |
这样我们能从他这几天跑步情况推断出这几天的天气。
在基因序列中也是同样的原理,只是由原先的时间关系变为了按碱基(氨基酸残基)顺序的关系
这样我们观测到的是一串碱基,但我们要推断出序列背后的含义。例如我们可以通过观测到有一串碱基,然后一算发现序列对应着TATA-box。当然这么做要现有一个对TATA-box的认识,也就是我们下载的HMM profile文件。
有人可能会疑惑,在上面的案例中,我们是一个状态(天气)对应一个观察(跑步),但是序列长度是不固定的,这又是怎么计算的呢?我本人水平有限,也没有想明白,如果有兴趣大家可以自己探索,参考推荐阅读部分的书。
下载并安装HMMER软件
这个软件需要从源码编译,windows系统下非常麻烦,建议使用linux系统。作为第一个要自己编译的软件,这里给一个步骤,后续要自己编译的软件都差不多。
试了一下,在windows下几乎无法完成,不用尝试了。
直接搜索可以找到对应的网站:http://hmmer.org/
下载完成后,解压
运行./configure
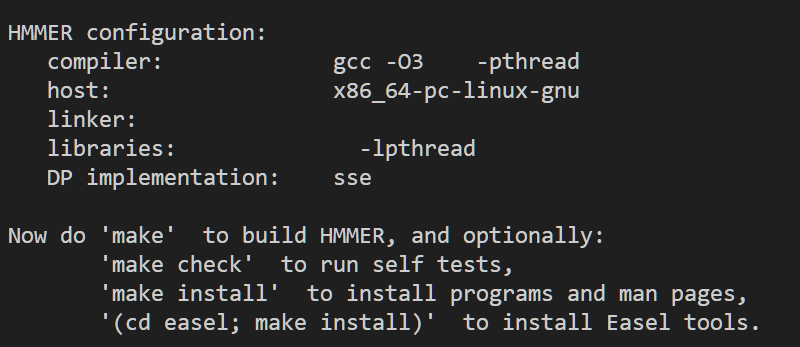
make install安装
等待结束就安装完成了
关于如何更改路径以及其它问题都应该看文档
查文献,下载hmm profile文件
hmmer是通过结构域来搜索的,所有我们要找到对应的描述文件
通过查文献的方式,看他们使用的是哪些文件。这一步应该在“阅读文献”步骤中完成,这里再重复一下
搜索相关的文章
按时间排序
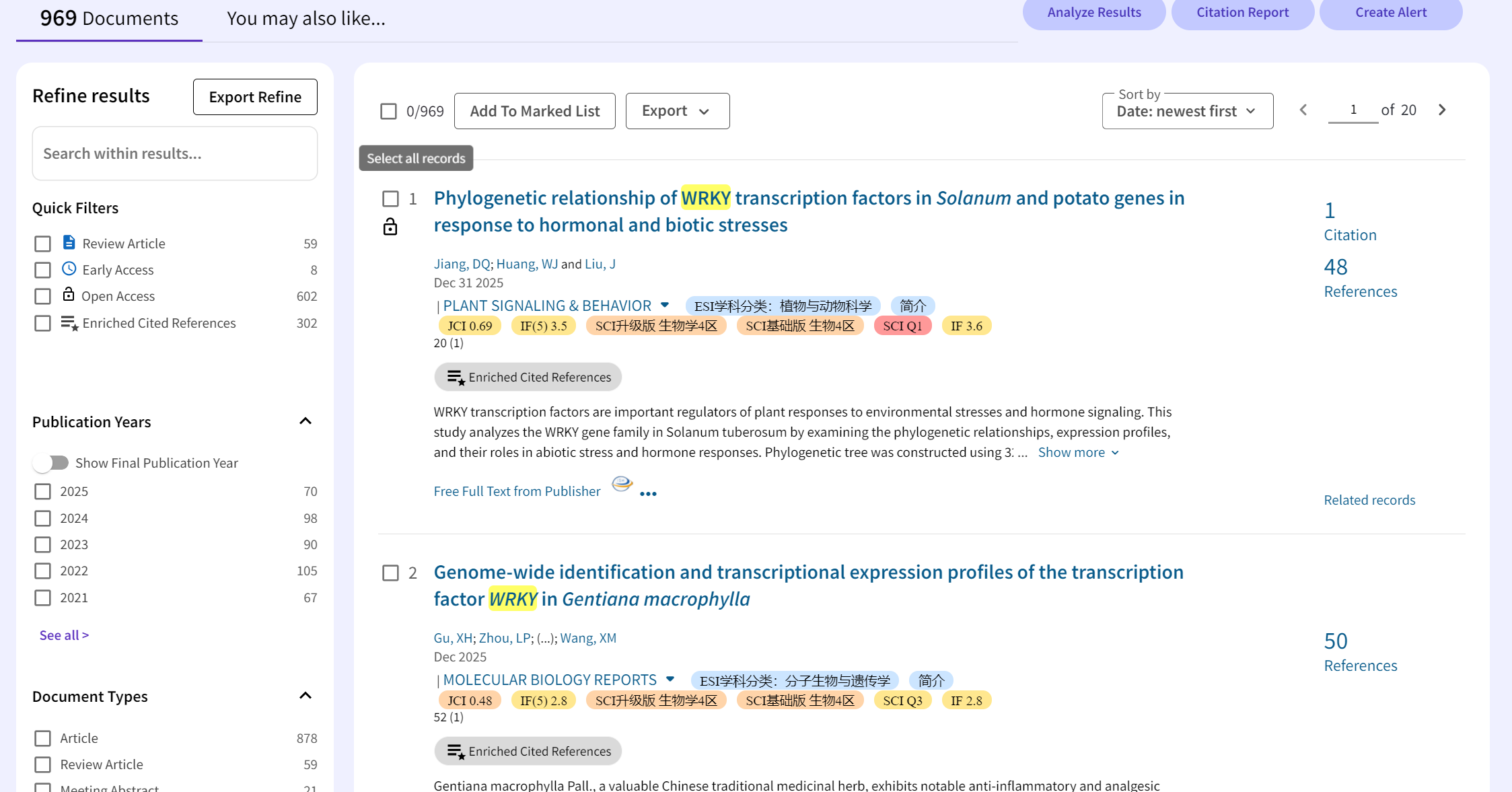

看到他们用了编号为:PF03106
打开interPro网站下载结构域的描述文件
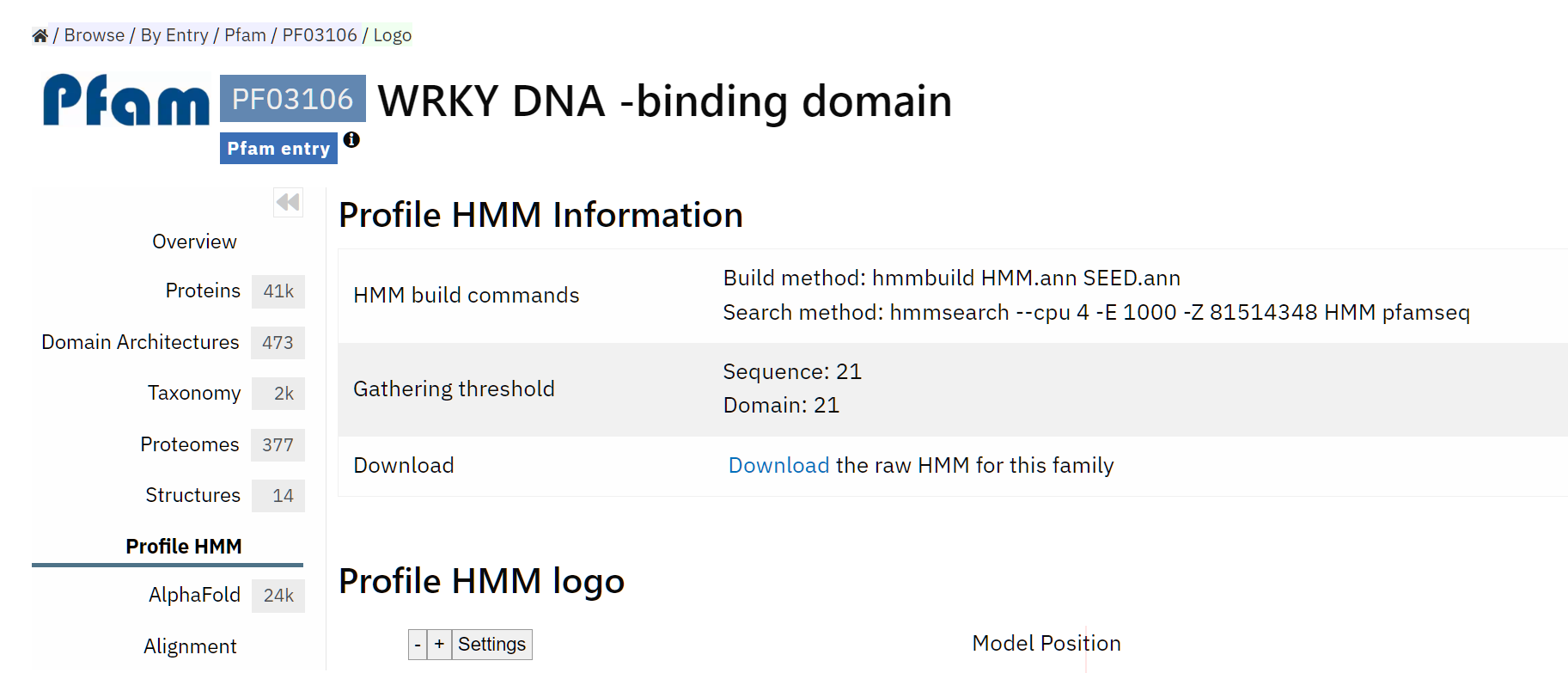
看到是WRKY binding domain,没错
下载下来就可以了
搜索序列
最完整的用法应该查文档
当然看文档很烦人,作者自己也不愿意看。文档中甚至还在教我们如何避免看文档


我们只需要知道怎么用就行了,用-h看一下
这里输入hmmsearch -o hmm_result.txt PF03106.hmm.gz wfs_pep.fa
结果长这样:
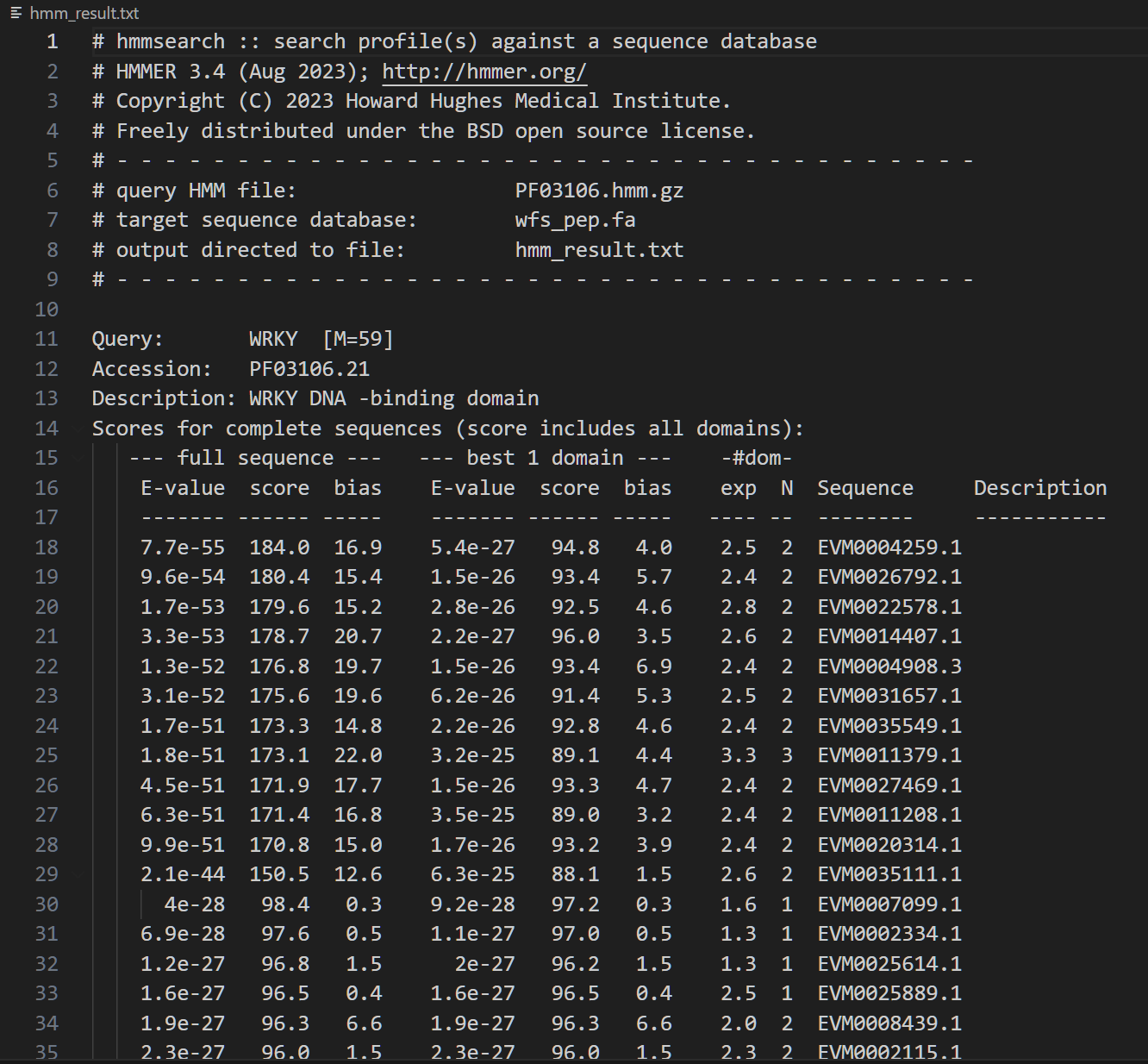
我们可以选择保留哪些
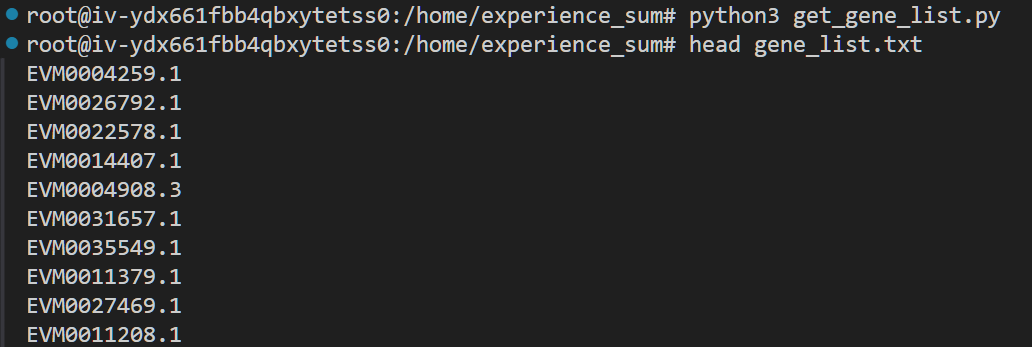
写一个简短的脚本提取这些基因编号
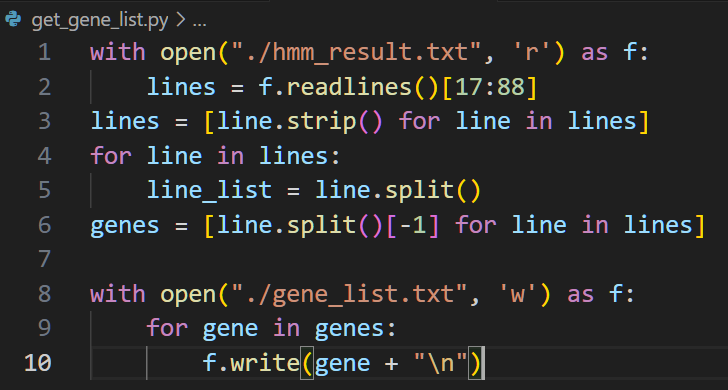
运行一下
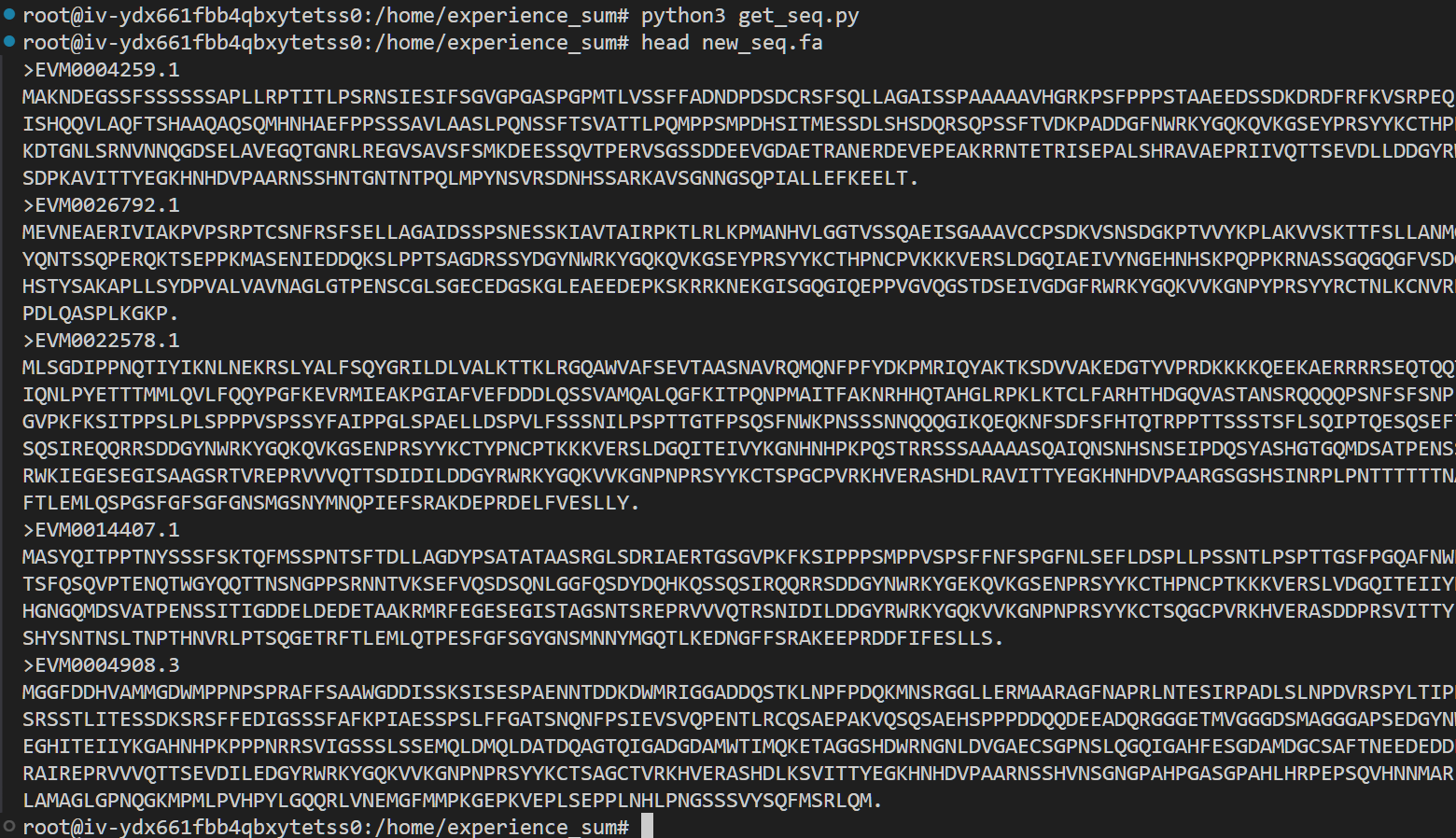
从fasta文件中提取序列
有了列表提取序列就很简单了,同样编写一个简单的脚本
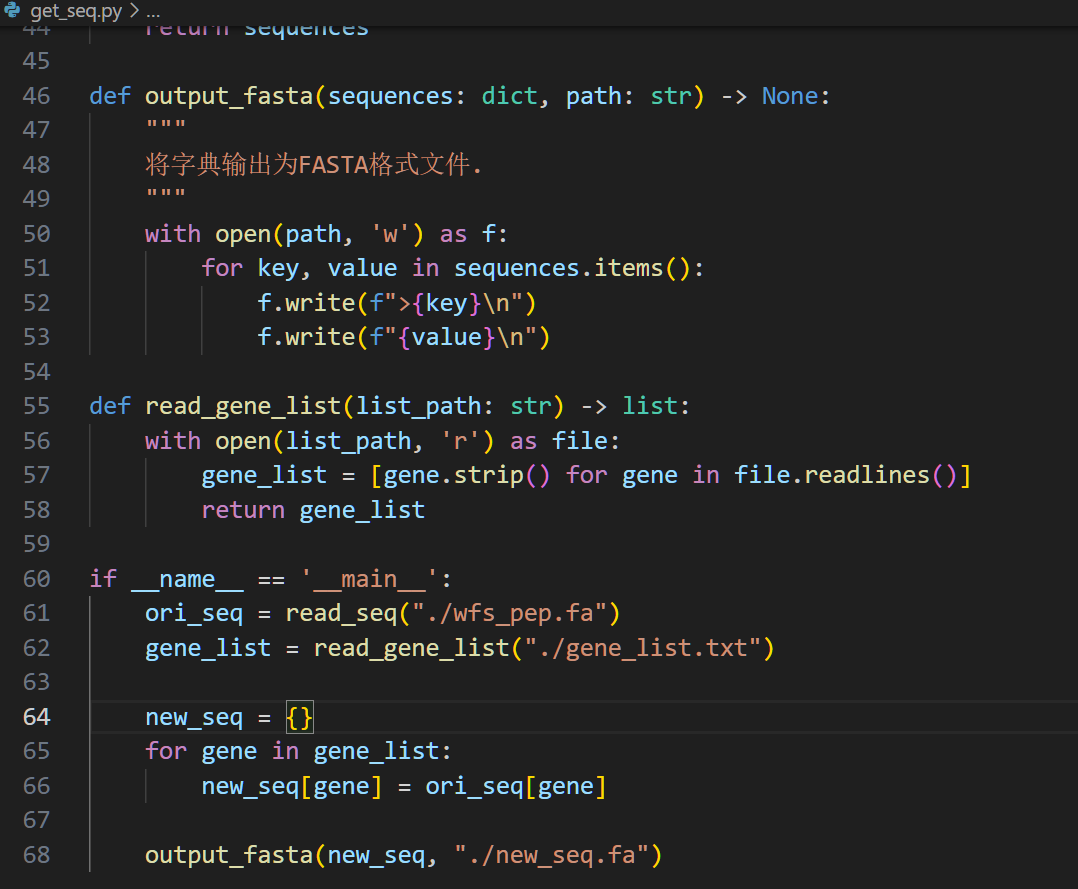
运行一下
推荐阅读
Biological sequence analysis, Richard Durbin et al., Cambridge University Press
系统发育树构建与绘制
系统发育树(Phylogenetic Tree)是研究物种进化关系的重要工具。它通过分析不同物种或基因之间的序列相似性来推断它们的进化历史。下面我们将详细介绍如何构建和绘制系统发育树。
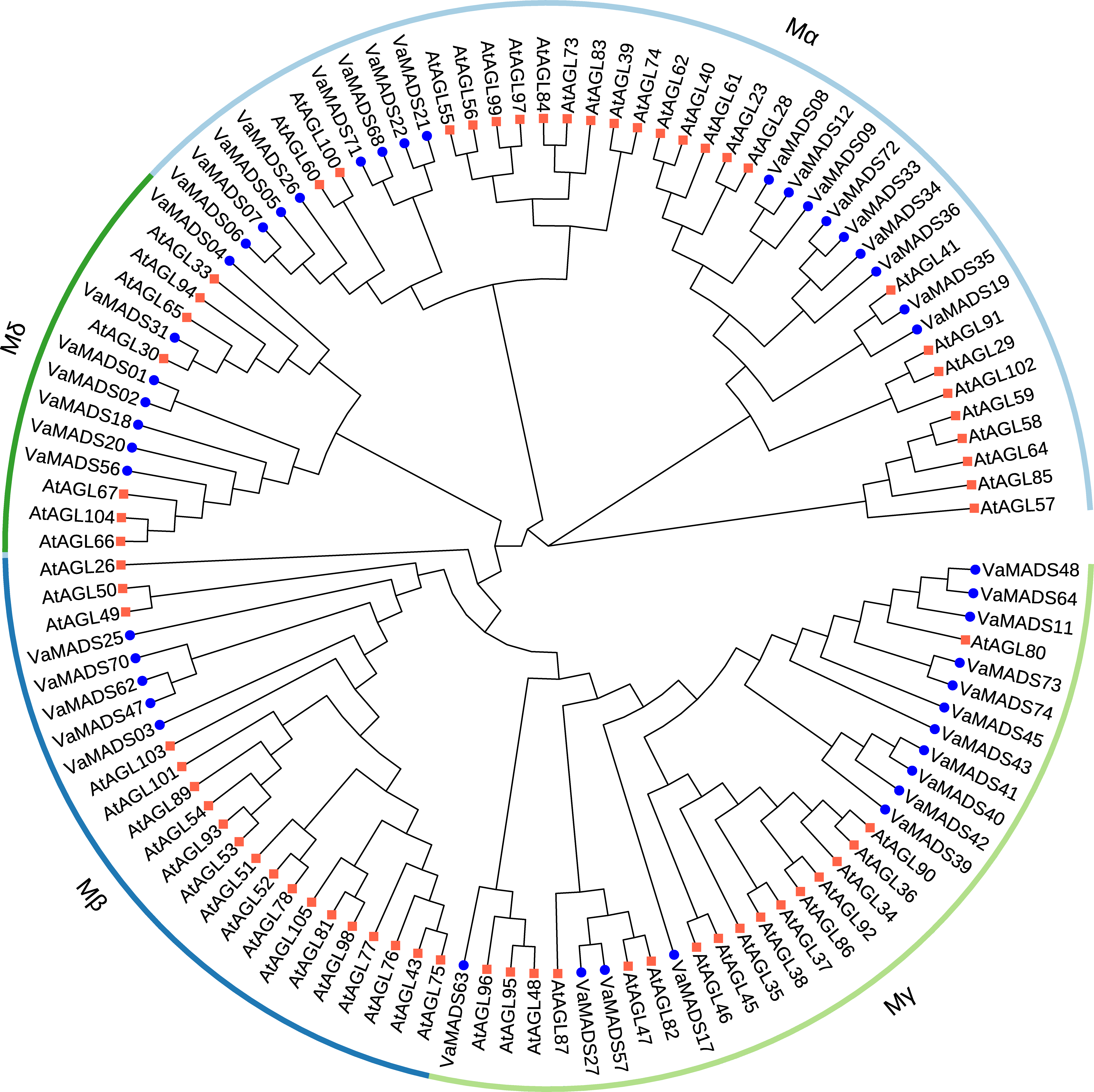
目标
- 构建一个系统发育树
- 对基因家族进行分类
- 绘制美观且易于理解的系统发育树图形
材料与软件
- MAFFT(多序列比对)
- R及ggtree包(绘制系统发育树)
- 蛋白质序列文件
主要步骤
- 对所有序列进行多序列比对
- 构建系统发育树
- 参考其它研究对基因进行分类
- 绘图
原理
系统发育树的构建基于分子数据(如DNA或蛋白质序列),通过比较这些序列之间的差异来估计它们之间可能存在的亲缘关系。常用的算法包括但不限于最大似然法(Maximum Likelihood, ML)、邻接法(Neighbor Joining, NJ)等。每种方法都有其优缺点,在实际应用时需根据具体情况选择最合适的一种。
邻接法(NJ)比较快,适用于大数据集。它根据序列间的遗传距离创建树形结构,通常作为初步探索性分析的一部分。
最大似然法(ML)这种方法虽然计算复杂度较高,但能够提供更为精确的结果。它假设存在一个最佳模型来解释观察到的数据,并尝试找到最符合该模型的树拓扑结构。
拟南芥参考序列的下载
我们可以在TAIR数据库中下载拟南芥的参考序列。但是自从2025年的某次更新后,TAIR不再能随意访问了,需要证明科研用途。有两个验证方法,一是注册一个账号(比较麻烦),二是使用校园网环境访问(现在只能用webvpn)。
找到了编号之后可以在这里批量下载:TAIR - Bulk Download - Sequences
webvpn这个东西很大概率没法通过机器人验证,反馈了也没有解决。没关系不影响TAIR的使用。
多序列比对
首先需要将所有感兴趣的序列进行多序列比对,以找出它们之间的同源区域。这一步骤对于确保系统发育树的准确性至关重要。
下载并安装MAFFT。
打开终端或命令行界面,输入以下命令:
其中input.fasta是准备好的序列文件,aligned.fasta则是输出的比对结果。
构建系统发育树
这里使用iqtree构建系统发育树,iqtree官网有各个平台的安装包。具体的使用看文档。
ClustalX也可以,有图形化界面一看就懂。
它们的建树方法不同,iqtree使用的是最大似然法,clustalx使用的是邻接法。
还有MEGA也可以建树,但是我已经遇到过不止三个人windows下的mega崩溃了,所以谨慎使用。
简单的示例:
-s 即为输入文件,支持很多格式
-nt 线程数
-pre 输出文件的前缀
输出如下:
.
├── example.phy
├── tree1.bionj
├── tree1.ckp.gz
├── tree1.iqtree
├── tree1.log
├── tree1.mldist
├── tree1.model.gz
└── tree1.treefile
其中treefile就是我们构建好的树
可视化系统发育树
使用R和ggtree包绘制系统发育树,ggtree的具体用法,应当自己查看文档。
如果不想用R,iTOL这个网站也可以绘图,有图形化的操作界面。
对基因进行分类
参考已有的研究,对不同分支的基因进行分类。一般来说都是按照分支进行划分,比如以拟南芥为参考进行划分。
Tips
在用iqtree建树时可能会遇到下面的情况:
显示gap过多可能会影响建树的结果。
这关于种情况本人查看了许多中文的英文的论坛。有些人认为gap本身也代表着进化的信息,有些人认为如果某个位点gap比例过大则可以直接删除这一列。
如果需要删除,这里介绍一个工具:trimAl
推荐阅读
《分子进化与系统发育》(美) Masatoshi Nei, Sudhir Kumar著 吕宝忠, 钟扬, 高莉萍等译。学校图书馆有
开心一下:
看这么长时间,应该也累了吧。
众所周知,康德是一位著名的哲学家,著有《纯粹理性批判》《实践理性批判》等。
康德在有一段时间在柯尼斯堡任教(就是那个七桥问题的柯尼斯堡)且生活十分有规律,每天准时下午3点半出门散步。以至当地的居民在他每天散步经过时来对表。但有一次康德在3点半没有出现,因为读卢梭的《爱弥儿》入迷,以致错过了散步的时间。结果导致当地的居民纷纷产生恐慌,在一个星期之内竟拿不定主意如何校对时间。
PS. 本人高中时买过商务印书馆出版的《纯粹理性批判》,想要拜读一下,可惜完全看不懂。但我认为不是我的问题,因为我翻了一下序言发现这个版本竟然是100多年前(清朝还在)翻译的,一整本半文言,真的看不懂。
共线性分析
基因组在演化过程中经历了许多不同的变异,包括基因组复制过程中的 DNA 突变、染色体重组、重复元件转座与插入、基因组及基因水平多倍化、横向基因转移等。
共线性分析的核心意义在于,它提供了一个宏观的、结构性的视角来解读基因组的演化历史和基因的功能关联。它通过考察基因在染色体上的排列顺序和邻里关系,为我们揭示了更大尺度上的进化事件。比如鉴定全基因组复制(WGD)和片段复制事件,能帮助我们重构物种演化过程中染色体的断裂、重组和融合等结构变异历史。更重要的是,保守的共线性关系是判断不同物种间基因是否为直系同源基因(因物种分化而来,功能更可能一致),这对于在新物种中准确预测基因功能、构建可靠的进化树以及开展比较基因组学研究至关重要。
选择压计算(计算Ka/Ks比值)是通过比较改变氨基酸的非同义突变(Ka)和不改变氨基酸的同义突变(Ks)的速率,我们可以直接推断一个基因所承受的进化压力类型。Ka/Ks < 1 意味着基因受到纯化选择,表明其功能至关重要,任何有害变异都会被清除(也就是能不变就不变);Ka/Ks = 1表明了中性选择;而 Ka/Ks > 1 则揭示了基因正在经历正选择,意味着新的突变带来了适应性优势,被自然选择所青睐。
目标
识别基因的共线性关系并计算选择压,绘图
材料与软件
- MCScanX(共线性识别软件)
- BLASTp
- KaKs_Calculator(选择压计算软件)
- PAL2NAL(多肽核酸相对应脚本)
- 蛋白质序列文件
- 核酸序列
- gff注释文件
- 还需要自己写一点程序处理数据
步骤
- 使用BLASTp对所有的蛋白质进行比对
- 利用MCScanX识别共线性
- 绘图
- 选择压计算
BLASTp
blast是一个双序列比对工具,将输入的序列与数据库中的序列进行对比,给出相似度得分
NCBI提供了在线的工具,当然我们要在基因组内部进行比较,所以不使用在线工具,需要自己本地运行。
安装
很简单,应该不用专门说明。
建库
显而易见,dbtype有两种(核酸和蛋白嘛)
比对
这里选择输出格式为6,会输出一张表格
输出文件一共有12列:
第1列: 输入序列的名称。
第2列: 比对到的目标序列名称。
第3列: 序列相似度。
第4列: 比对的有效长度。
第5列: 错配数。
第6列: gap数。
第7-8列: 输入序列比对上的起始和终止位置。
第9-10列: 比对到目标序列的起始和终止位置。
第11列: e-value。
第12列: 比对得分。
Tips
因为是对全基因组进行比较,所以会略微耗时
MCScanX搜索
这个软件需要从源码编译
用法
MCScanX dir/xyz
这里要注意,blast的结果要命名为xyz.blast,gff注释文件也要有相同的文件名xyz.gff否则软件无法识别。
结果有三个:xyz.collinearity、xyz.tandem和一个文件夹xyz.html
其中collinearity是共线性结果,tandem为片段重复,html文件里输出了每条染色体上的共线性情况。


由于这是对全基因组的筛查,所以我们还需要自己编写程序,将它处理以下。
概念
共线性在英文里有许多说法st和collinearity。概念上略有区别。
其中可以分为片段重复(segmental duplication)和串联重复(tandem duplication)。
串联重复的概念似乎就是该软件的作者提出的。基因在染色体上的距离小于200kb就为串联重复。
串联重复和片段重复在进化上的功能不同,具体可以查阅相关书籍。
绘图
R包circlize可以完成这种环形图的绘制,具体用法也要查看该包的文档。
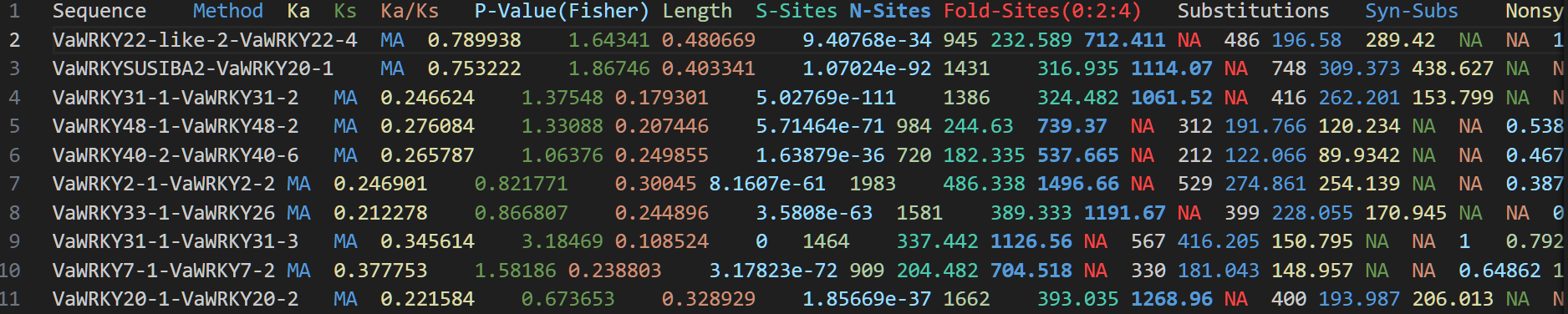
选择压计算
Ka(非同义突变率):指基因序列中导致氨基酸改变的突变率。
Ks(同义突变率):指基因序列中不改变氨基酸的突变率。
Ka/Ks 比值:通过比较 Ka 和 Ks,判断基因是否受到选择压力。
Ka/Ks比值的含义:
- Ka/Ks < 1:纯化选择(负选择),表明非同义突变对生物体有害,被自然选择淘汰。
- Ka/Ks = 1:中性进化,表明非同义突变和同义突变以相同速率积累,不受选择压力影响。
- Ka/Ks > 1:正选择,表明非同义突变对生物体有利,被自然选择保留。
计算
这一步需要我们自己写许多脚本,相当麻烦。
显然我们需要先找到核酸,然后才能计算它们的突变,所以要使用PAL2NAL找到蛋白质对应的核酸序列。
用法很简单,使用-h看一下帮助就会了。
这样我们就有了外显子的核酸序列
这里使用KaKs_Calculator计算,该软件有Windows版本,而且有图形界面。
该软件需要一种独特的文件格式.axt作为输入。
格式如下:
VaWRKY70-2-VaWRKY70-3
ATGGACTCCTCCACTTCGCCGGAGAACTTATTCGGTGATCGGAAC...
ATGGACAATTCC---TCGCCGGAGAACTTGTTCGGTGACCGGAAC...
VaMADS01-VaMADS56
---------------------ATGAAAAAGATAGAGAACTTAACA...
ATGGGTCGTGTTAAGCTTCAAATAAGGAAGATAGAAAACACGACA...
一行基因对的名称和两行序列
所以这里需要我们自行编写程序
- 将共线性基因对从全基因组的结果中筛出来
- 将它们的蛋白质序列找到
- 每对(两条)序列进行多序列比对
- 根据原始的核酸序列找到蛋白序列对应的核酸(使用pal2nal)
- 合并这些结果,并处理为KaKs_Calculator可用的格式
然后就可以计算了
推荐阅读
《植物基因组学》樊龙江著,ISBN:9787030633132
开心一下:
这一部分可以说是最麻烦的了,因为需要我们自己做相当多的工作。如果你做完了,恭喜你,可以放松一下。
因为谈恋爱弄丢了自己老爹诺奖的小故事(道听途说,不保真):
众所周知,DNA的双螺旋是沃森(Watson)和克里克(Crick)发现的。但是有一位大佬也差一点提前发现了该结果,他就是两次独立获得诺贝尔奖、化学键理论的提出者鲍林(Pauling)。鲍林利用自己提出的化学键理论,成功计算出了α-螺旋的结构。在成功计算出α-螺旋的结构后,他也有兴趣进一步探究DNA的结构。当罗莎琳德·富兰克林(Rosalind Frankling, 这也是一位大佬,只可惜去世得比较早,不然也能拿奖)拍出了DNA的X射线衍射图谱之后,鲍林也想去看一看。以他的水平绝对一看就能立马明白这是一个双螺旋,但当时他的年纪大了,于是他就派他的儿子去那边看看。但是他的儿子竟然迷上了富兰克林的一位女助手,天天想着怎么和她发展关系,于是他不仅没有完成他父亲交给他的打探消息的任务,还把这边的情况给透露了。在这期间,沃森和克里克成功拿到了该结果,并且对其进行了分析,最终成功提出了DNA双螺旋的结构(然后我们都知道,得奖了)。
这个故事告诉我们工作要专心。



基因结构分析
众所周知,基因不是直接翻译为蛋白质的,含有内含子外显子等,所以我们要把它绘制出来。有什么用呢?一是看到结构上的差异,这种差异可以支持我们的预测和猜想。例如我们之前将基因进行了分组,现在发现同组基因的结构相似,证明了我们分组的正确性;对于不同组,我们发现有的组内含子多,有的组几乎没有,验证了一定进化上的关系。
motif和domain是处于二级结构和三级结构中间的一种结构,有的生物化学书或是专业词典上对它们有明确的定义。我觉得最重要的是认识到domain一般比较长,有定名,有一定的独立的功能;而motif较短,是对一组序列进行分析得出的共有的相似的序列,所以没有固定的名称。在实际分析中我们可以观察到一些整齐出现的motif,这时可以猜测这些motif可能组成了某些domain因而有一定的作用。
目标
绘制基因外显子内含子结构图和表示motif的结构图

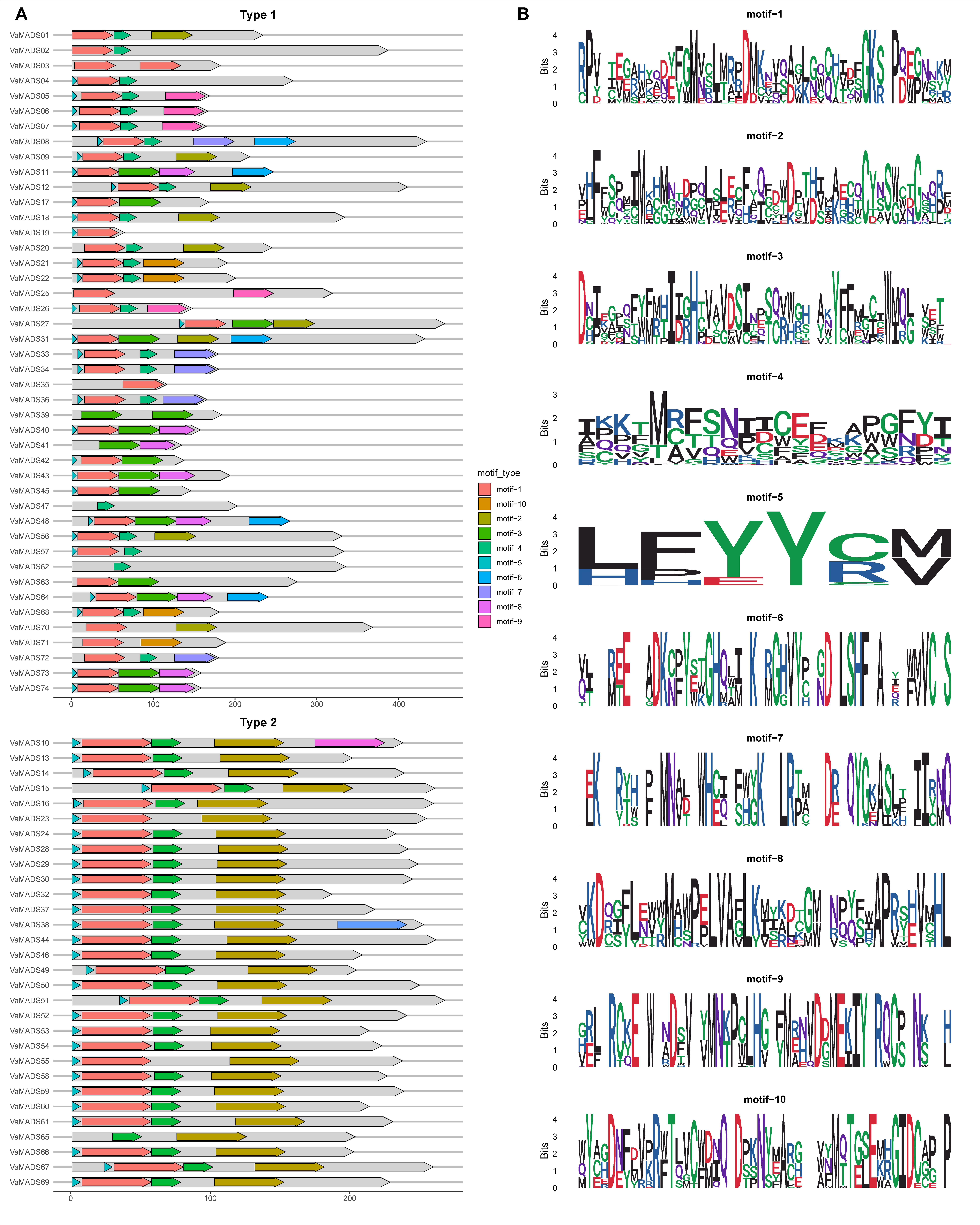
材料与软件
- gff注释文件
- R及gggenes包
- gsds网站(绘制内含子外显子,也可以用R绘制,我还没有尝试)
- MEME suit(motif预测,按道理来说motif只是一段重复出现的相似度比较高的序列,自己写一个查找软件也是可以的)
主要步骤
- 处理现有数据,上传gsds绘图
- 上传序列至MEME-suit,下载结果
- 处理结果数据,绘图
基因结构绘制
这一步相当简单,看一下网站上的格式要求,简单用R处理一下就可以了。甚至可能不需要处理,直接用基因家族的gff注释就可以了。
Motif分析
很不巧,写着一部分的时候meme在维护,无法展示图片。
不过同样也很简单,上传一下序列就可以了。下载格式应该有xml格式和txt格式
我们可能需要对xml进行解析
待完善
绘图
然后用gggenes绘制就可以了,Logo图用ggseqlogo,文档需要自己看。数据处理为什么格式,也要自己根据绘图来调整
推荐阅读
R for Data Science, 2nd edition, Hadley Wickham
数据科学中的R语言,王敏杰
顺式作用元件
我们都知道,顺式作用元件就是对基因有调控功能的一段序列,位于基因上游。它们和反式作用因子结合调控基因转录。种类有启动子、增强子、沉默子等。通过对顺式作用元件的分析,我们可以预测被其调控的基因参与了哪些生物学过程。
目标
绘制一幅图
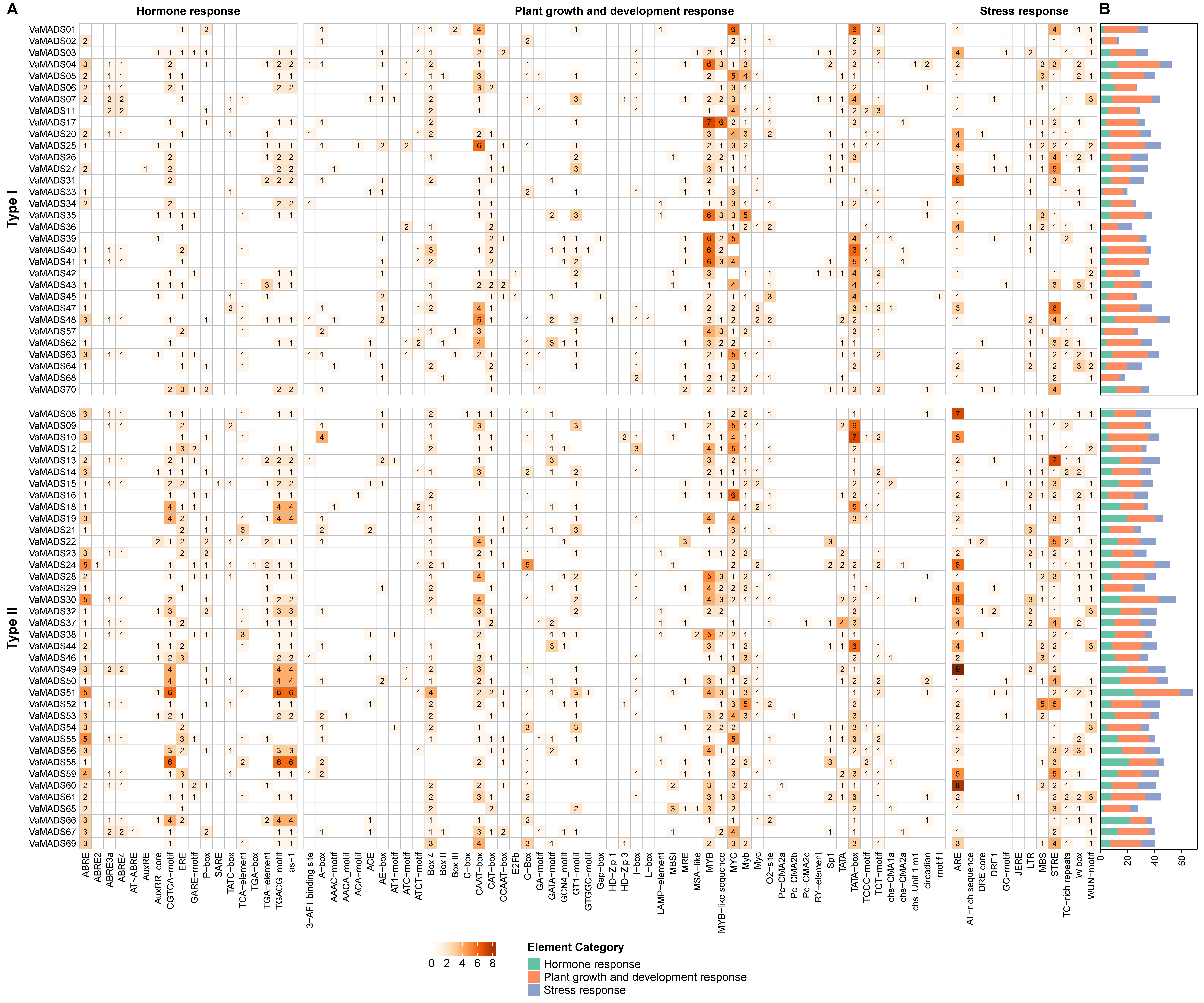
这里我用一幅热图表示分析结果,有的文章里会将顺式作用元件在上游的位置表示出来,我觉得热图更能说明情况。
材料与软件
- 基因组文件
- 基因组注释文件
- PlantCARE网站(搜索顺式作用元件)
- R及complexheatmap包(绘制热图)
- 自己写一个小程序提取上游序列
主要步骤
- 从注释文件中找到基因的起始位点
- 提取起始位点上游2000bp序列
- 提交至plantcare分析并下载结果
- 整理数据,绘图
找到起始位点并提取上游序列
这一步可以说是最麻烦的,因为我们要自己实现:读取并解析gff注释->找到该基因的起始位点->读取序列文件->找到上游的序列->提取上游序列
这也是锻炼我们编程水平
PlantCARE分析
推荐阅读
开心一下:
之前提到过柯尼斯堡七桥问题,后来欧拉把它抽象为图论的数学问题,然后得出了不能一次走完结论(即不能一笔画)。但我认为作为一个现实问题,七桥问题还是有特殊解的,就是沿河而上,跨过河流的源头再返回,OK了。
基因表达分析
这里值得注意,"expression pattern" 和 "expression profile" 都叫做“表达模式”,但是通过阅读文章,profile似乎表示处理上的不同(也就是分为对照组和实验组,进行不同的处理),pattern表示时间上的不同(不同发育阶段)。
这一步可以通过绘图来观察出不同基因的表达。
目标
绘制热图
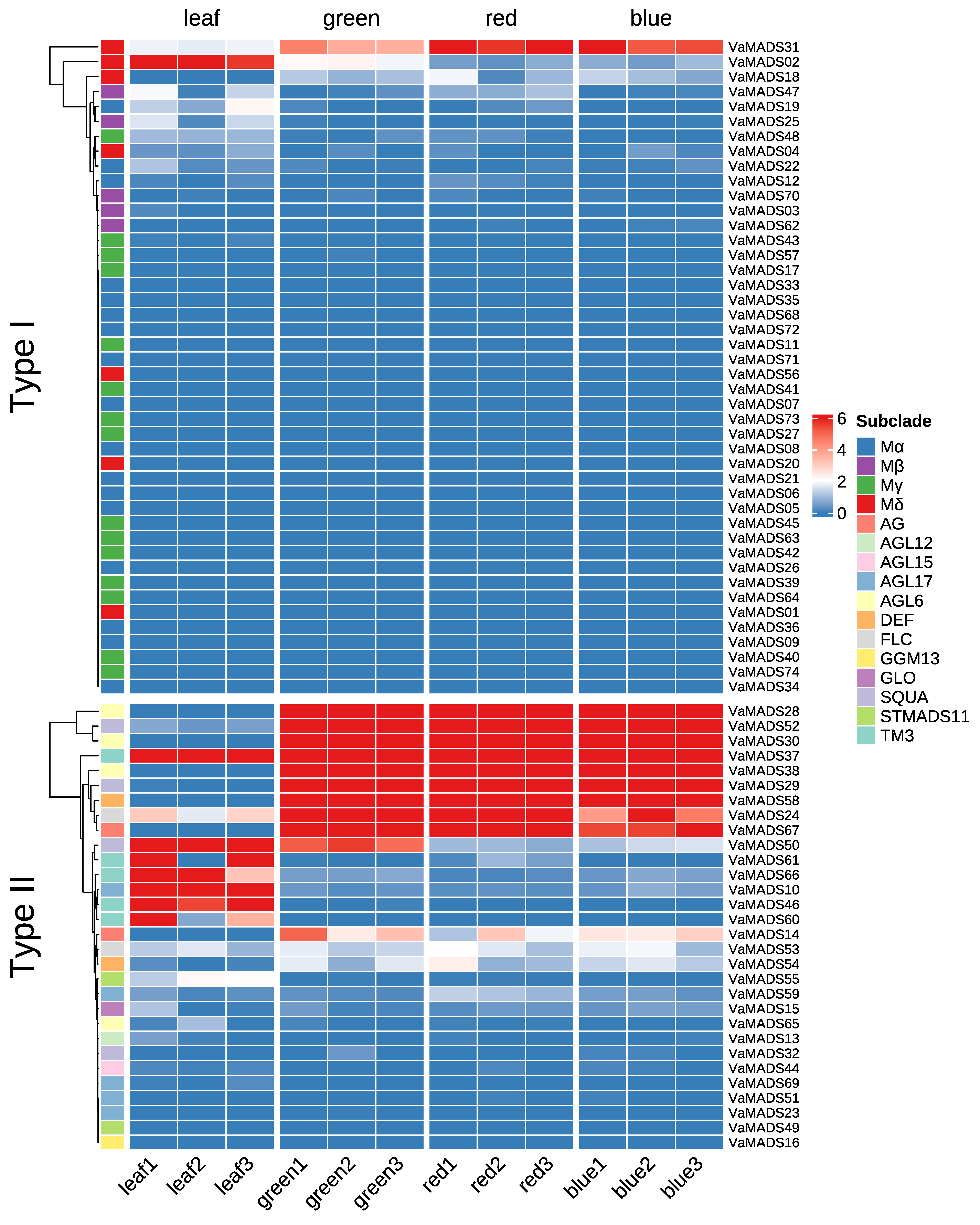
这个任务用Excel也能完成,很简单。
蛋白质互作
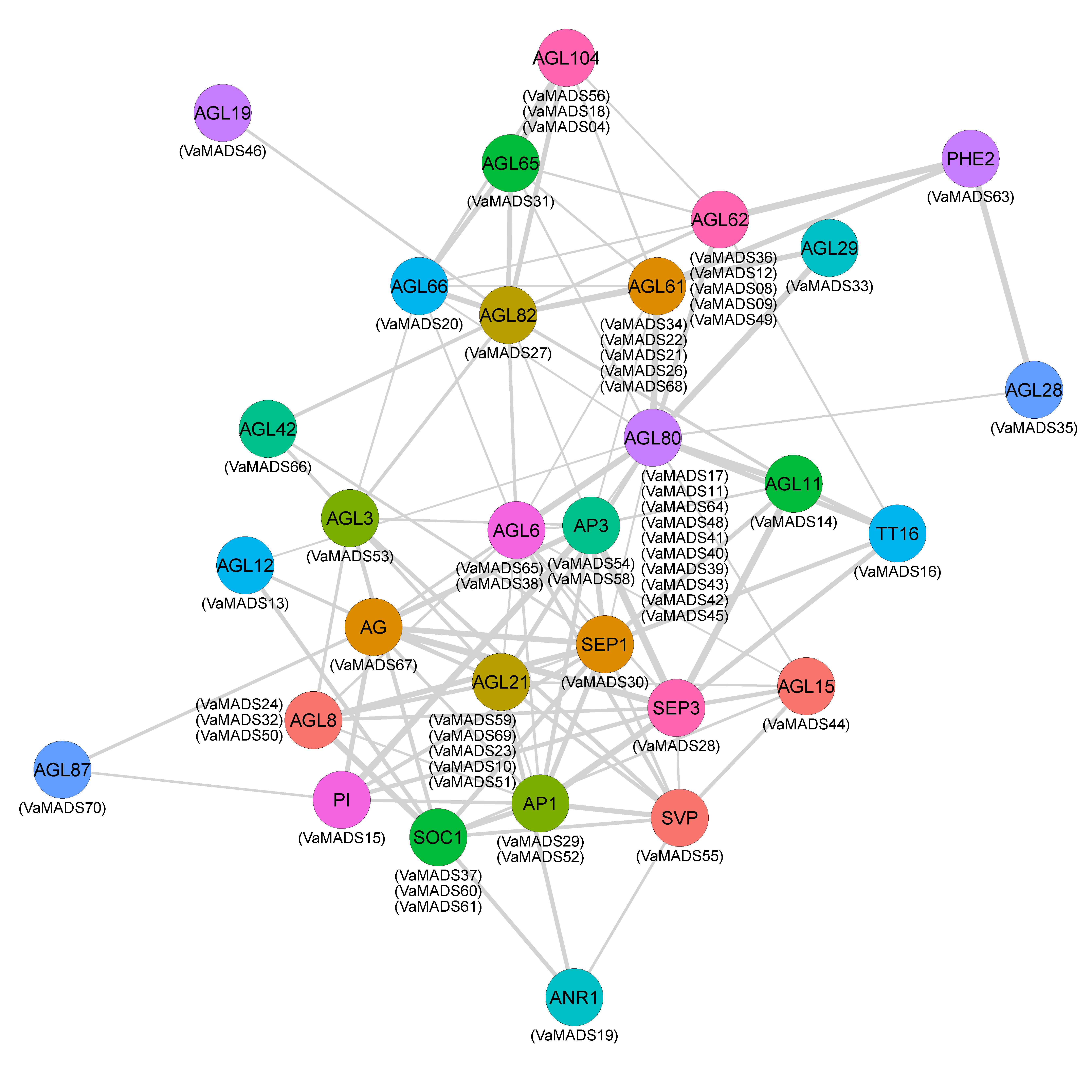
蛋白质-蛋白质相互作用(PPI)网络是一种用于描绘一组蛋白质之间相互作用关系的分析方法。由于蛋白质间的相互作用关系错综复杂,每个蛋白质被视为网络中的一个节点,而蛋白质之间的相互作用则通过连线来表示,从而形成了复杂的网状结构。
PPI网络分析通过识别在PPI网络中占据关键位置的蛋白质,可以为生物标志物的筛选提供重要的理论依据和技术支持。这些关键蛋白往往在细胞功能或信号传导过程中发挥着核心调控作用,因此它们对于理解生物过程和疾病机理具有重要价值。
目标
构建目标基因家族的蛋白质互作网络,并通过可视化分析识别其中的关键互作关系和核心蛋白。
材料与软件
- 基因家族的蛋白质序列文件(fasta格式)
- STRING 数据库(用于预测蛋白质相互作用)
- R及ggraph包
主要步骤
- 获取蛋白列表
- 在STRING数据库中进行预测
- 导出网络文件
- R进行绘图
- 分析网络拓扑结构
获取蛋白列表
整理出我们在第一步“识别并找出基因家族的基因”中鉴定出的所有蛋白质的ID列表或其fasta序列。
在STRING数据库中进行预测
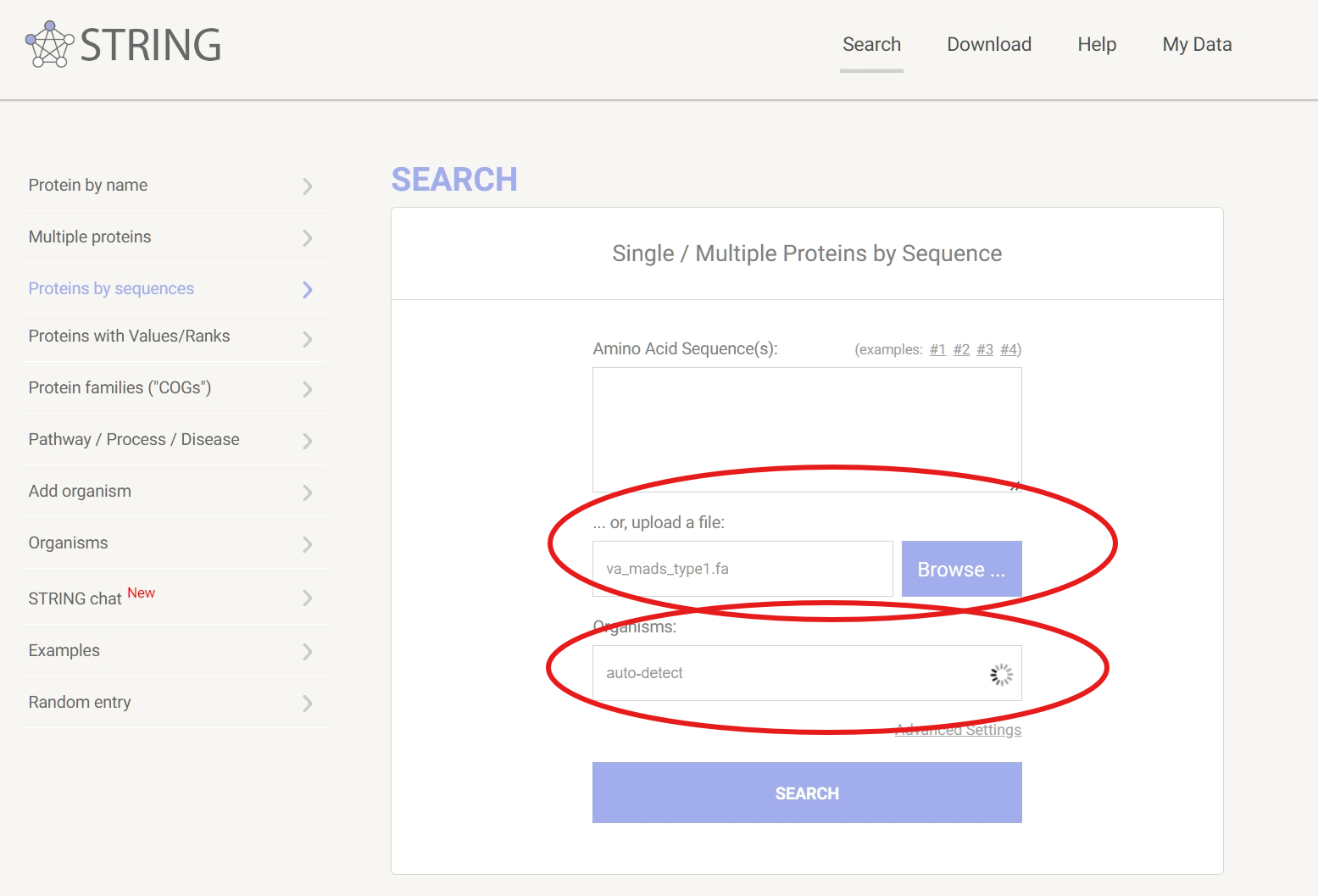
访问STRING官网,将我们的蛋白质序列粘贴到“Multiple proteins”的输入框中或是直接上传序列文件。选择一个参考物种,然后点击“Search”。
STRING预测的原理可能是对蛋白质进行比对,如果发现数据库有相似性高的蛋白质,则参考已有的数据完成预测。STRING会整合来自实验数据、文本挖掘、共表达分析等多种来源的证据,返回一个预测的互作网络图。string输出的图中的连线有不同的颜色,代表不同的证据来源;连线的粗细代表了相互作用的置信度得分(confidence score)。
导出网络文件
为了进行更自由的美化和深入分析,我们需要将网络数据导出。在STRING的结果页面,切换到“Exports”标签页,下载TSV格式或cys.json格式的网络文件。TSV文件包含了互作的蛋白对和置信度得分,格式简单,通用性强。
绘制网络图并分析
R中的ggraph包可以完成这个任务,具体使用需要看文档。STRING数据库也有一个官方的软件cytospace,也可以完成绘制。
推荐阅读
要真正搞懂网络图,恐怕还要学点图论,但是我不会。